

Alpha-1-Antitrypsin Deficiency: Embracing Awareness
Alpha-1-antitrypsin deficiency is a codominant hereditary condition that impacts the lungs and liver, presenting various challenges in medical practice. This genetic disorder affects the production of alpha-1-antitrypsin protein, leading to an imbalance of protease activity and subsequent tissue damage. The perspectives on treating alpha-1-antitrypsin deficiency encompass a multidimensional approach aimed at alleviating symptoms, slowing disease progression, and enhancing the overall quality of life for affected individuals. Since the only treatment method is inept, some emerging therapeutic approaches offer hope for improved management of alpha-1-antitrypsin deficiency.
Alpha-1-antitrypsin
Alpha-1-antitrypsin (A1AT), otherwise called alpha-1 proteinase inhibitor, is a water-soluble glycoprotein abundantly found in a healthy human’s bloodstream. Produced mainly by hepatocytes, this member of the serpin superfamily plays a crucial role in the homeostasis of connective tissues. Other sources of A1AT include monocytes, alveolar macrophages, neutrophils and type II pneumocytes. A1AT is encoded by the SERPINA1 gene. It possesses more than a hundred allelic variants resulting in different amounts of produced A1AT in distinct individuals (Pini et al., 2021). This protease inhibitor has a blood range reference of 0.9–2.3 g/L, although its levels increase during inflammation. Proteins whose concentrations rise or fall rapidly (by at least 25% in plasma) in response to inflammatory stimuli are called ‘acute-phase proteins’ (APP). A1AT is classified as a positive APP, which means that its serum concentration increases under the influence of cytokines, such as IL-1, IL-6 or TNFα (Ackermann, 2017). Weighing 52 kDa, it is both an endogenous and an exogenous protease inhibitor. The term ‘antitrypsin’ was coined by researchers when they first discovered its ability to bind and irreversibly inactivate trypsin (Brebner & Stockley, 2013). Misleadingly, A1AT’s primary function is inhibiting not trypsin but neutrophil elastase (NE). NE partakes in various immunological processes, such as degrading phagocytised microorganisms or inducing cytokines and chemokines and is usually found inside neutrophils. However, it appears on the outside of cells during inflammation or infection, acting as an essential component of neutrophils’ extracellular traps. Uninhibited, NE initiates proteolytic degradation; therefore, the production of fully functional A1AT is needed to maintain the balance between proteases and antiproteases. The caused deterioration of the extracellular matrix in airways prompts airflow obstruction by weakening the elasticity of the bronchial walls (M. Hunt & Tuder, 2012).
However, A1AT performs numerous other functions. Alongside NE, it inhibits other human neutrophil serine proteases, notably cathepsin G and proteinase 3. When these enzymes bind to A1AT, its reactive centre loop is destroyed, which results in the degradation of both the protease and A1AT. It is worth noting that A1AT plays more of a balancing role instead of a nullifying one, as it maintains the required level of neutrophil proteases necessary for host defence while simultaneously granting protection to alveoli (Janciauskiene et al., 2018).
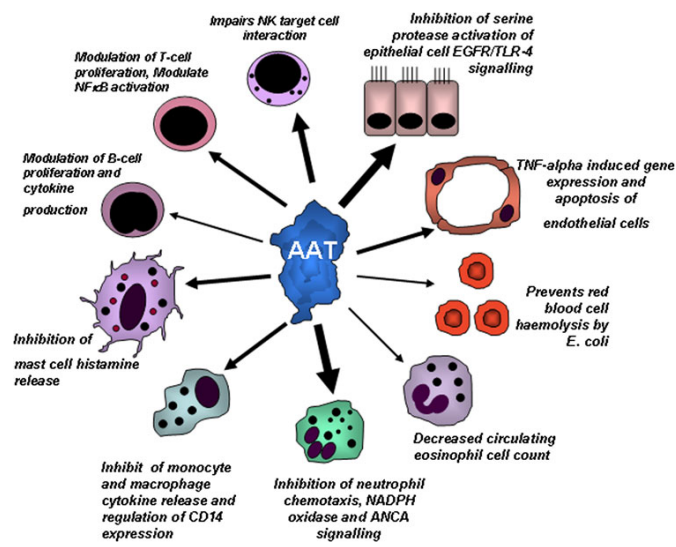
Recent studies have revealed that A1AT takes part in anti-inflammatory and immunomodulatory processes. Admittedly, this protein defends cells against apoptosis, interrupts the production of superoxides by neutrophils, regulates the expression of specific cytokines and enzymes, and inhibits non-serine proteases (Janciauskiene, 2017). It also can enhance insulin-induced mitogenesis and stop the adhesion of neutrophils to fibrinogen (Al-Omari et al., 2011). Finally, it plays a role in iron metabolism by regulating the synthesis of ferritin and transferrin receptors (Janciauskiene, 2017). Among other things, A1AT also interacts with adaptive immunity mechanisms, affecting the function of Th1, Th2, and Th17 T cells and inducing the generation of T-regulatory cells (Tregs).
A1AT displays extensive genetic variation. The mutations give rise to various phenotypes categorised based on the protein’s migration in an isoelectric gradient. Examples include PiA (anodal variants), PiM (normal), PiS (slow), and PiZ (slowest). The PiZ mutation is of particular clinical significance, being the leading cause of severe α-1-antitrypsin deficiency (AATD) (M. Hunt & Tuder, 2012).
Alpha-1-antitrypsin deficiency
Ever since its discovery in 1963, α-1-antitrypsin deficiency remains a severely underdiagnosed condition. This rare autosomal codominant hereditary condition affects 1 in 2000 Western Europeans with a gradual decline to the East. The vast majority of those affected are Caucasian, with little to no cases recorded in Southeast Asia and Africa (de Serres & Blanco, 2014). Mutations in the SERPINA1 gene result in a decreased level of A1AT (Perkins et al., 2017).
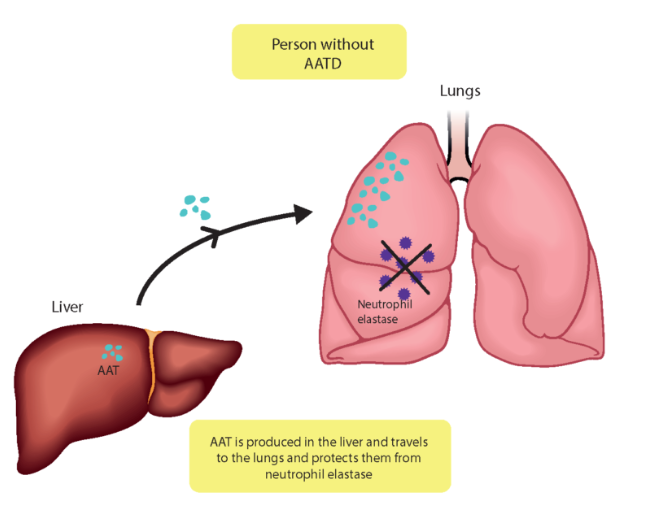
Principally, AATD has pulmonary and hepatic manifestations and, consequently, leads to the development of symptoms and signs characteristic of chronic hepatitis, cirrhosis, and chronic obstructive pulmonary disease (COPD) (Rodrigues et al., 2020). That includes emphysema, bronchiectasis, and chronic bronchitis (Hatipoğlu & Stoller, 2016). COPD is a group of respiratory tract inflammatory diseases generally characterised by disturbances in the expiratory gas flow leading to its accumulation in the lungs (Bals, 2010). Many scientists strongly underline the fact that AATD remains under-recognised. One of the reasons might be that the associated symptoms are often linked to exogenous causes, such as cigarettes (Bouchecareilh, 2014). Indeed, smoking has been proven to accelerate pathological lesions in AATD, although, unfortunately, non-smoking individuals with AATD still develop COPD, even though it occurs at an older age (Strange, 2020). Emphysema, for instance, is a condition characterised by the destruction of the bronchi walls, overproduction of mucus, and loss of physiologically active alveoli. As a result, it becomes impossible to properly evacuate all mucus, which turns into a nest for bacteria and cytokines, promoting and exacerbating inflammatory processes in the lungs (Janssen et al., 2019).

Apart from the lungs, hepatocytes, where A1AT is produced chiefly, are also prone to deterioration. Thanks to the periodic acid–Schiff–diastase stain, researchers have determined the cause of AATD-associated liver damage. The polymerisation and improper folding of the A1AT protein block its secretion, induce proteotoxic stress and lead to hepatocytes injury, finally resulting in the development of hepatitis followed by cirrhosis. This pathological accumulation of A1AT has also been linked to liver carcinogenesis (Clark, 2017).
Prevention and Treatment
Unfortunately, there are no preventive measures that could be implemented since the disorder is genetically acquired. Given the hereditary nature of the disease, a family screening is recommended (Hatipoğlu & Stoller, 2016). Some scientists list quitting smoking as a preventive measure, although that action can only delay lung destruction (Miravitlles et al., 2017). Oftentimes, in patients suffering from AATD, multiple forms of COPD coexist, which additionally impedes therapeutic choices. As is the case with many disorders, early detection significantly boosts the chances of combatting the effects of AATD (Torres-Durán et al., 2018). A study by M Luisetti and N Seersholm has shown that, perplexingly, barely 0.35% of predicted cases in Canada and Europe have been diagnosed. The cause for it is that patients often do not exhibit any symptoms in the early stages or show signs that bear a resemblance to other, more known conditions, such as non-alcoholic steatohepatitis, liver failure, asthma or bronchiectasis (Luisetti & Seersholm, 2004).
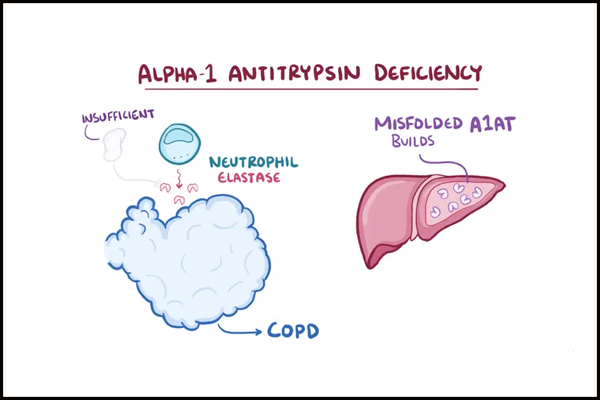
For patients with severe AATD, only augmentation therapy has been approved, even though it raises some controversies regarding its efficacy. Augmentation therapy is the gold-standard treatment option for patients with severe lung disease. It consists of periodic intravenous injections of pooled human plasma enriched with A1AT. Although augmentation therapy reduces the frequency of infections in the respiratory system, it is not without disadvantages (Kelly et al., 2010). For example, it fails to halt the process of lung injury. Furthermore, the weekly necessity of getting intravenous injections is both expensive and highly inconvenient. Finally, concerns about the risk of transmitting infections arose and led to the exploration of transgenic and recombinant A1AT. Admittedly, A1AT was successfully obtained with the help of bacteria, yeast and transgenic sheep. Nonetheless, the produced recombinant proteins were either undergoing abnormal glycosylation or wholly lacking it, which made them unusable (Lorincz & Curiel, 2020).
Another approach implements a nebulizer, which permits an effective delivery of the desired compound directly to the alveoli. The major challenge of this method is transferring enough A1AT to the interstitium without it being stuck on the alveoli epithelial wall. Even without considering the convenience aspect, the method’s advantage lies within a potentially higher concentration of particles being distributed precisely to the lungs, as barely 2 to 3% of A1AT given intravenously reaches them (Brebner & Stockley, 2013).

In some cases, lung transplantation is also available. Advances in surgical techniques and perioperative care have significantly increased the survival rate of the procedure. Even so, it remains a considerable life-threatening challenge, as an abundance of complications can arise, among others – primary graft dysfunction, chronic lung allograft dysfunction or opportunistic infection. The last one is a direct result of immunosuppressive therapy, which is indeed imperative in post-operational management but carries the risk of contagion and developing tumours (Meyer, 2018). Overall, lung transplantation has been proven to prolong the patient’s life expectancy. Patients with AATD, who have survived at least eight years after surgery, died of entirely unrelated causes (Stone et al., 2016). When it comes to the liver, the only remedy is liver transplantation. Sadly, it can lead to severe complications and even death from haemorrhage, thrombosis, infection, severe graft dysfunction or chronic rejection (Stoller et al., 2020).
Stem cell therapy is one of the most prominent tools in regenerative medicine. Plentiful research has been conducted using host-induced pluripotent stem cells (Duvoix et al., 2014). In a study by Rashid et al. (2010), patient-specific hepatocytes were developed from previously procured dermal fibroblasts from AATD individuals. Immunostaining and ELISA analyses have confirmed that the hepatocytes have replicated the key features of the disease, as A1AT polymer accumulation has been observed. Even though previous studies have already proven that the entrapment of polymers within the ER is the underlying cause of the clinical phenotype, the variations in the phenotype among those with the exact genotype remain unclear. Each individual probably has a different capability for dealing with protein aggregations (Rashid et al., 2010). Another study by Wilson et al. showed an approach that harnesses the ability of hematopoietic stem cells to expand in vivo and uses lentiviruses to integrate genes into their genome. Despite having accomplished the transduction of the majority of the hematopoietic system, it did not result in a circulating A1AT concentration above the so-called ‘protective threshold’ [800 mg/L in human serum], the level thought to prevent the development of emphysema. Nonetheless, the authors encourage further research, as they believe that the transduction of a small number of stem cells might be able to achieve a sustained A1AT expression in the future (Wilson et al., 2008).
One of the emerging perspectives in treating A1AT deficiency is the potential use of mRNA-based therapies. mRNA, or messenger RNA, has gained significant attention in recent years as a versatile tool in medicine. In the context of AATD, mRNA-based treatments hold promise in addressing the underlying genetic defect responsible for the condition. This innovative approach involves delivering synthetic mRNA molecules that encode the blueprint of the A1AT protein into the patient’s cells. Once inside the cells, the production of the missing or defective protein begins. This technology has shown encouraging results in preclinical studies and is being explored in clinical trials for various genetic disorders. However, the availability, cost, and accessibility of this novel treatment remain factors that influence its widespread use.
In theory, cellular transplants capable of secreting A1AT may be engrafted into AATD patients’ tissues. Still, notable challenges remain, particularly delivering the necessary quantities of A1AT capable of protecting the lung parenchymal tissues. Lastly, a concern remains that the transplanted cells could proliferate and rival the native stem cell populations (McNulty et al., 2021).
Conclusions
Treating AATD presents various obstacles. From a clinical standpoint, early diagnosis and intervention are crucial to prevent or slow down the progression of the disease. Therapeutic strategies for managing AATD range from lifestyle modifications, such as quitting smoking and environmental exposure reduction, to pharmacological interventions. The only treatment option focuses only on slowing down the disease's progress. Thankfully, approaches like gene therapy and mRNA-based therapy hold promise in addressing the underlying genetic defect and potentially providing long-term benefits.

Public knowledge of AATD is minimal, contributing to challenges in early detection, diagnosis, and appropriate management of the condition. AATD is considered a rare genetic disorder, which often means it receives less attention and awareness than more prevalent diseases. As a result, many individuals, including healthcare professionals, may have limited familiarity with AATD, its symptoms, and associated risks. Comprehensive patient care requires a multidisciplinary approach involving pulmonologists, hepatologists, genetic counsellors, and respiratory therapists to optimise the management and support of individuals with A1AT deficiency. The combination of medical advancements, personalised care, and patient education might enhance the quality of life for those affected by this complex genetic disorder.
Bibliographical References
Ackermann, M. R. (2017). Inflammation and Healing. Pathologic Basis of Veterinary Disease Expert Consult, 73-131.e2. https://doi.org/10.1016/B978-0-323-35775-3.00003-5
Al-Omari, M., Korenbaum, E., Ballmaier, M., Lehmann, U., Jonigk, D., Manstein, D. J., Welte, T., Mahadeva, R., & Janciauskiene, S. (2011). Acute-phase protein α1-antitrypsin inhibits neutrophil calpain I and induces random migration. Molecular Medicine (Cambridge, Mass.), 17(9–10), 865–874. https://doi.org/10.2119/molmed.2011.00089
Bals, R. (2010). Alpha-1-antitrypsin deficiency. Best Practice and Research: Clinical Gastroenterology, 24(5), 629–633. https://doi.org/10.1016/j.bpg.2010.08.006
Bouchecareilh, M. (2014). Alpha-1-antitrypsin deficiency. In Medecine/Sciences (Vol. 30, Issue 10, pp. 889–895). Editions EDK. https://doi.org/10.1051/medsci/20143010016
Brebner, J. A., & Stockley, R. A. (2013). Recent advances in α-1-antitrypsin deficiency-related lung disease. In Expert Review of Respiratory Medicine (Vol. 7, Issue 3, pp. 213–230). Expert Reviews Ltd. https://doi.org/10.1586/ers.13.20
Clark, V. C. (2017). Liver Transplantation in Alpha-1 Antitrypsin Deficiency. In Clinics in Liver Disease (Vol. 21, Issue 2, pp. 355–365). W.B. Saunders. https://doi.org/10.1016/j.cld.2016.12.008
de Serres, F., & Blanco, I. (2014). Role of alpha-1 antitrypsin in human health and disease. In Journal of Internal Medicine (Vol. 276, Issue 4, pp. 311–335). Blackwell Publishing Ltd. https://doi.org/10.1111/joim.12239
Duvoix, A., Roussel, B. D., & Lomas, D. A. (2014). Molecular pathogenesis of alpha-1-antitrypsin deficiency. In Revue des Maladies Respiratoires (Vol. 31, Issue 10, pp. 992–1002). Elsevier Masson SAS. https://doi.org/10.1016/j.rmr.2014.03.015
Hatipoğlu, U., & Stoller, J. K. (2016). α1-Antitrypsin Deficiency. In Clinics in Chest Medicine (Vol. 37, Issue 3, pp. 487–504). W.B. Saunders. https://doi.org/10.1016/j.ccm.2016.04.011
Janciauskiene, S. (2017). Novel Biological Functions of A1AT. In Alpha-1-antitrypsin Deficiency: Biology, Diagnosis, Clinical Significance, and Emerging Therapies. Elsevier Inc. https://doi.org/10.1016/B978-0-12-803942-7.00006-4
Janciauskiene, S., Wrenger, S., Immenschuh, S., Olejnicka, B., Greulich, T., Welte, T., & Chorostowska-Wynimko, J. (2018). The multifaceted effects of Alpha1-Antitrypsin on neutrophil functions. Frontiers in Pharmacology, 9(APR), 1–13. https://doi.org/10.3389/fphar.2018.00341
Janssen, R., Piscaer, I., Franssen, F. M. E., & Wouters, E. F. M. (2019). Emphysema: looking beyond alpha-1 antitrypsin deficiency. Expert Review of Respiratory Medicine, 13(4), 381–397. https://doi.org/10.1080/17476348.2019.1580575
Kelly, E., Greene, C. M., Carroll, T. P., McElvaney, N. G., & O’Neill, S. J. (2010). Alpha-1 antitrypsin deficiency. In Respiratory Medicine (Vol. 104, Issue 6, pp. 763–772). W.B. Saunders Ltd. https://doi.org/10.1016/j.rmed.2010.01.016
Lorincz, R., & Curiel, D. T. (2020). Advances in alpha-1 antitrypsin gene therapy. In American Journal of Respiratory Cell and Molecular Biology (Vol. 63, Issue 5, pp. 560–570). American Thoracic Society. https://doi.org/10.1165/RCMB.2020-0159PS
Luisetti, M., & Seersholm, N. (2004). a1-Antitrypsin deficiency 1: Epidemiology of a1-antitrypsin deficiency. 164–169.
M. Hunt, J., & Tuder, R. (2012). Alpha 1 Anti-Trypsin: One Protein, Many Functions. Current Molecular Medicine, 12(7), 827–835. https://doi.org/10.2174/156652412801318755
McNulty, M. J., Silberstein, D. Z., Kuhn, B. T., Padgett, H. S., Nandi, S., McDonald, K. A., & Cross, C. E. (2021). Alpha-1 antitrypsin deficiency and recombinant protein sources with focus on plant sources: Updates, challenges and perspectives. In Free Radical Biology and Medicine (Vol. 163, pp. 10–30). Elsevier Inc. https://doi.org/10.1016/j.freeradbiomed.2020.11.030
Meyer, K. C. (2018). Recent advances in lung transplantation [version 1; peer review: 2 approved]. F1000Research, 7(0), 1–11. https://doi.org/10.12688/F1000RESEARCH.15393.1
Miravitlles, M., Dirksen, A., Ferrarotti, I., Koblizek, V., Lange, P., Mahadeva, R., McElvaney, N. G., Parr, D., Piitulainen, E., Roche, N., Stolk, J., Thabut, G., Turner, A., Vogelmeier, C., & Stockley, R. A. (2017). European Respiratory Society statement: Diagnosis and treatment of pulmonary disease in α1-antitrypsin deficiency. European Respiratory Journal, 50(5). https://doi.org/10.1183/13993003.00610-2017
Perkins, J. T., Choate, R., Mannino, D. M., Browning, S. R., & Sandhaus, R. A. (2017). Benefits Among Patients with Alpha-1 Antitrypsin Deficiency Enrolled in a Disease Management and Prevention Program. Chronic Obstructive Pulmonary Diseases: Journal of the COPD Foundation, 4(1), 56–64. https://doi.org/10.15326/jcopdf.4.1.2016.0161
Pini, L., Paoletti, G., Heffler, E., Tantucci, C., & Puggioni, F. (2021). Alpha1-antitrypsin deficiency and asthma. In Current Opinion in Allergy and Clinical Immunology (Vol. 21, Issue 1, pp. 46–51). Lippincott Williams and Wilkins. https://doi.org/10.1097/ACI.0000000000000711
Rashid, S. T., Corbineau, S., Hannan, N., Marciniak, S. J., & Miranda, E. (2010). Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. Conflict, 120(9), 3127–3136. https://doi.org/10.1172/JCI43122DS1
Rodrigues, J. F., Mineiro, A., Reis, A., Ventura, D. G., Fernandez-Llimos, F., Costa, F., Gomes, J., Silva, J. M., Lopes, P., & Cordeiro, C. R. (2020). Alpha-1 antitrypsin deficiency: Principles of care. Acta Medica Portuguesa, 33(6), 433–439. https://doi.org/10.20344/amp.12950
Stoller, J. K., Lacbawan, F. L., & Aboussouan, L. S. (2020). Alpha-1 Antitrypsin Deficiency Summary Diagnosis Suggestive Findings. U.S. National Library of Medicine, 1–27.
Stone, H. M., Edgar, R. G., Thompson, R. D., & Stockley, R. A. (2016). Lung transplantation in alpha-1-antitrypsin deficiency. COPD: Journal of Chronic Obstructive Pulmonary Disease, 13(2), 146–152. https://doi.org/10.3109/15412555.2015.1048850
Strange, C. (2020). Alpha-1 Antitrypsin Deficiency Associated COPD. In Clinics in Chest Medicine (Vol. 41, Issue 3, pp. 339–345). W.B. Saunders. https://doi.org/10.1016/j.ccm.2020.05.003
Torres-Durán, M., Lopez-Campos, J. L., Barrecheguren, M., Miravitlles, M., Martinez-Delgado, B., Castillo, S., Escribano, A., Baloira, A., Navarro-Garcia, M. M., Pellicer, D., Bañuls, L., Magallón, M., Casas, F., & Dasí, F. (2018). Alpha-1 antitrypsin deficiency: Outstanding questions and future directions. In Orphanet Journal of Rare Diseases (Vol. 13, Issue 1). BioMed Central Ltd. https://doi.org/10.1186/s13023-018-0856-9
Wilson, A. A., Kwok, L. W., Hovav, A. H., Ohle, S. J., Little, F. F., Fine, A., & Kotton, D. N. (2008). Sustained expression of α1-antitrypsin after transplantation of manipulated hematopoietic stem cells. American Journal of Respiratory Cell and Molecular Biology, 39(2), 133–141. https://doi.org/10.1165/rcmb.2007-0133OC
Visual Sources
Cover Image: [Illustration showing a diseased human liver]. (n.d., 2022). [Illustration]. Medthority. https://www.medthority.com/specialty/gastroenterology-and-hepatology/alpha-1-antitrypsin-deficiency-aatd-liver-disease/
Figure 1: The many functions of A1AT. Bergin, D.A., Hurley, K., McElvaney, N.G. et al. Alpha-1 Antitrypsin: A Potent Anti-Inflammatory and Potential Novel Therapeutic Agent. Arch. Immunol. Ther. Exp.60, 81–97 (2012). [Illustration]. https://doi.org/10.1007/s00005-012-0162-5
Figure 2: A1AT protects a healthy person's lungs. (n.d., 2021). [Illustration]. European Lung Foundation. https://europeanlung.org/en/information-hub/lung-conditions/alpha-1-antitrypsin-deficiency/
Figure 3: A1AT is unable to perform its function in a person with severe AATD. (n.d., 2021). [Illustration]. European Lung Foundation. https://europeanlung.org/en/information-hub/lung-conditions/alpha-1-antitrypsin-deficiency/
Figure 4: In AATD, both the lungs and the liver suffer. (Chauhan, 2020). [Illustration]. Planet Ayurveda. https://www.planetayurveda.net/what-is-alpha-1-antitrypsin-deficience-can-we-manage-it/
Figure 5: The various approaches for treating AATD. (Goel, 2021). [Illustration]. Touch Respiratory. https://www.touchrespiratory.com/copd/journal-articles/updates-in-the-management-of-alpha-1-antitrypsin-deficiency-lung-disease/#article
Figure 6: Specific recommendations for treating patients with AATD in the intensive care unit. (Rahaghi, 2019). [Illustration]. Rahaghi, F., Omert, L., Clark, V., & Sandhaus, R. A. (2019). Managing the Alpha-1 patient in the ICU: Adapting broad critical care strategies in AATD. Journal of Critical Care, 54, 212–219. https://doi.org/10.1016/j.jcrc.2019.08.027
Figure 7: An advertisement spreading awareness about AATD. (n.d.). [Illustration]. Alpha-1 Association of Australia. https://www.alpha1.org.au/index.php/63-announcements/88-november-is-alpha-1-awareness-month


Comments